







Thérapie génique et ophtalmologie

Grâce à de nouveaux travaux menés à l’Institut de la Vision la promesse des thérapies géniques - restaurer la vision en corrigeant les erreurs du code génétique - se rapproche un peu plus de la réalité. L’utilisation de modèles d’organes humains permettrait d’améliorer leur efficacité et sécurité, pour l’ophtalmologie et même au delà. Entretien avec la première auteure de l’article scientifique qui offre ces espoirs concrets.

Dr Juliette Pulman,
Post-doctorante dans l’équipe Thérapie géniques et modèles des maladies neurodégénératives, dirigée par Deniz Dalkara, à l’Institut de la Vision.
Et si l’on pouvait tester les thérapies géniques des maladies de la rétine chez l’humain avant leur administration aux patients ? C’est le projet sur lequel travaille le Dr Juliette Pulman, post-doctorante à l’Institut de la Vision, et ses collègues de l’équipe Thérapie géniques et modèles des maladies neurodégénératives. Leurs travaux, publiés en décembre 2025 dans le journal Molecular Therapy : Methods & Clinical Development, montrent que l’utilisation des organoïdes rétiniens, des modèles de rétines humaines, pourraient être le chaînon manquant pour améliorer les essais cliniques de thérapie génique par la technique CRISPR-Cas9.
Des ciseaux moléculaires de haute précision
En 2020, la française Emmanuelle Charpentier et l’américaine Jennifer Doudna reçoivent le prix Nobel de Chimie pour une technologie de « chirurgie de l’ADN » : CRISPR-Cas9. Ce système est en effet capable de cibler et de modifier des séquences très précises au sein du génome. Il repose sur l'interaction entre deux molécules : une enzyme, la protéine Cas9, capable de couper l’ADN, et un ARN guide (sgRNAs), une courte séquence synthétique, complémentaire de la séquence génétique cible. Celui-ci est conçu pour être le « GPS » du système. Ces deux éléments essentiels forment un complexe dit ribonucléoprotéique (RNP) qui « scanne » l’ADN jusqu’à ce que le sgRNA reconnaisse sa cible et s’y fixe. La conformation de la double hélice d’ADN, spécifique à chaque cellule différenciée, joue un rôle sur la capacité de CRISPR/Cas9 à s’y fixer. Une fois la fixation effectuée, l’enzyme Cas9 provoque une cassure double brin de l’ADN à cette localisation. La cellule met ensuite en marche ses mécanismes de réparation, mais le résultat est souvent imparfait, ce qui va provoquer des erreurs qui viennent désactiver le gène auquel appartient la séquence cible.
En tirant partie de ces erreurs, il est donc possible d’utiliser ce système pour corriger ou désactiver des gènes responsables de maladies spécifiques. C’est ce qu’ont prouvé Charpentier et Doudna il y a maintenant une dizaine d’année. Or, les maladies rétiniennes héréditaires, comme la rétinite pigmentaire, sont souvent causées par des mutations dans des gènes spécifiques. Elles sont donc de bonnes candidates pour des thérapies géniques via CRISPR-Cas9. Juliette Pulman et son collègue Hugo Malki, se sont concentrés sur le gène RHO, qui code pour la rhodopsine. Ce pigment visuel est présent dans les photorécepteurs, ces cellules sensibles à la lumière, est essentiel à la vision nocturne. Certaines des mutations touchant ce gène provoquent une accumulation de rhodopsine qui finit par être toxique pour les photorécepteurs, provoquant leur dégénérescence dans une forme de rétinopathie pigmentaire pour laquelle il n’existe pas de traitement à l’heure actuelle.
(B) Organoïde rétinien de 150 jours, la présence de cils à sa surface indique la présence de photorécepteurs.
(C) Cryosection d’un organoïde rétinien. En rouge la rhodopsine, indicatrice de la présence de bâtonnets ; en vert l’arrestine, indicatrice de la présence de cônes.
Modèles animaux versus modèles humains
Jusqu’à présent, pour tester l’efficacité et la sureté des stratégies thérapeutiques, les chercheurs dépendaient principalement de modèles cellulaires in vitro et de modèles animaux des pathologies. Mais dans le cas des approches par édition de gène avec la technologie CRISPR-Cas9, ces modèles sont insuffisants. En effet, « les cultures cellulaires couramment utilisée dans les études sur les thérapies Dr Juliette Pulman géniques sont des cellules en division. Leur ADN n’est pas du tout conformé comme celui des cellules matures, différenciées, présentes dans l’œil humain. Elles ne sont donc pas un bon modèle pour des thérapies d’édition de gènes in vivo » détaille Juliette Pulman. D’autre part, l’un des défauts de la technologie CRISPR est le risque de provoquer des coupures double brin de l’ADN à des endroits « hors cible ». Les modèles animaux ne sont donc pas non plus la réponse : comme ils ne présentent pas la même génétique que les patients humains, ils ne permettent pas de tester la sécurité de l’approche par CRISPR-Cas9 pour les pathologies humaines.
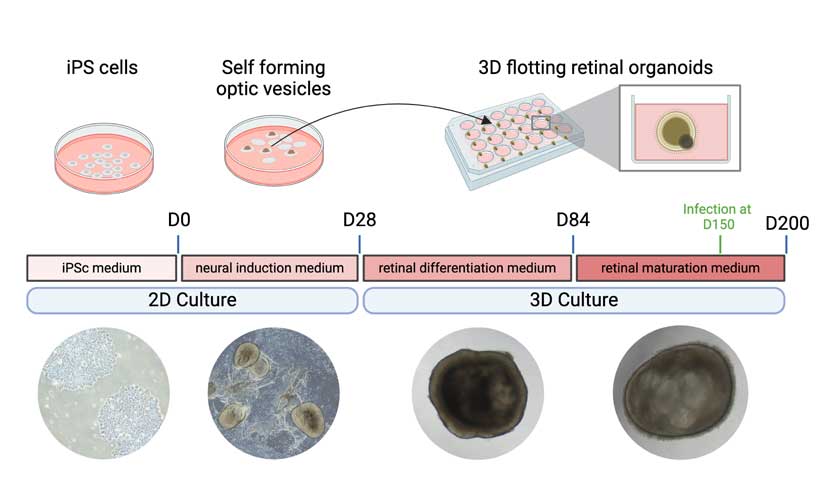
Ce qu’ont cherché à démontrer Juliette Pulman et son collègue dans leur publication, c’est qu’un autre type de modèle, dérivé de cellules humaines, pourrait permettre d’optimiser les thérapies géniques ciblant les photorécepteurs. Ce modèle, ce sont des organoïdes rétiniens, de minuscules « boules de cellules d’un demi millimètre, cultivées en laboratoire à partir de cellules souches humaines et capables de s’autoorganiser en 3D pour reproduire la différenciation et l’architecture de la rétine humaine » précise la chercheuse. Même si ceux-ci ne reproduisent pas l’ensemble des caractéristiques de la rétine, il leur manque notamment la vascularisation, leur développement à partir de cellules souches permet de récapituler le développement embryonnaire normal. A 150 jours de maturité, ces « mini-rétines » ont donc développé des structures naturellement présentes dans la rétine, notamment des photorécepteurs fonctionnels dont le segment externe capte la lumière. Autre avantage de taille, ces organoïdes « offrent un contexte génétique humain, permettant une évaluation bien supérieure de l’efficacité des sgRNAs et surtout du risque de coupure « hors cible » lors de l’utilisation de CRISPR Cas9. »
Comparer les réponses à la thérapie génique
Juliette Pulman a donc testé l’efficacité de l’outil d’édition génétique CRISPR Cas9 sur ces organoïdes, en ciblant spécifiquement le gène RHO, impliqué dans la rétinite pigmentaire autosomique dominante. Son objectif ? Comparer les résultats obtenus dans les organoïdes à ceux observés in vivo, chez la souris. Le tout en cherchant à obtenir une expression de la protéine Cas9 dans le noyau des cellules traitées qui soit transitoire plutôt que définitive, afin de limiter les risques de coupure «hors cible ».
La chercheuse a d’abord identifié, grâce à un criblage in vitro, des ARN guides (sgRNAs) capables de cibler efficacement le gène RHO. Elle a ensuite délivré ces outils d’édition génétique aux organoïdes rétiniens et aux rétines de souris humanisées, porteuses de mutations sur le gène RHO. Deux méthodes de vectorisation ont été comparées : la diffusion directe des ribonucléoprotéines (RNP) de Cas9, et la vectorisation via des vecteurs viraux AAV (Adeno-Associated Virus), couramment utilisés en thérapie génique pour leur capacité à infecter spécifiquement les photorécepteurs. Quelle que soit la méthode de vectorisation choisie, les taux d’édition génétique observés dans les organoïdes se sont révélés proches de ceux obtenus in vivo chez la souris. Preuve que « les organoïdes rétiniens reflètent mieux les conditions in vivo que les cellules en culture, où l’efficacité d’édition est bien plus élevée mais moins prédictive [des résultats in vivo de la thérapie génique] » commentent les auteurs dans la publication.

Dr Juliette Pulman
Vers une médecine personnalisée pour les maladies rétiniennes
Les implications de ces travaux sont majeures pour les ophtalmologistes et leurs patients. En effet, dans le domaine des maladies rétiniennes il y a, à l’heure actuelle, un grand nombre d’essais cliniques de thérapie par édition de gène en cours. Mais nombreux sont ceux qui échouent dès la phase 1, qui doit prouver leur efficacité et leur sécurité. Le transfert des résultats obtenus sur modèle animal est en effet difficile chez l’homme. En rajoutant une étape de validation chez l’humain avant le passage in vivo chez les patients, les organoïdes rétiniens pourraient accélérer le développement des traitements. Ils pourraient notamment permettre un criblage rapide et précis des ARN guides les plus efficaces pour cibler une mutation donnée ; une évaluation précoce de la toxicité des vecteurs et des outils d’édition génétique ; ou encore une meilleure prédiction des résultats cliniques, en évitant les biais liés aux modèles animaux. « Les organoïdes pourraient devenir un standard pour le développement préclinique des thérapies géniques, réduisant les risques pour les patients et augmentant les chances de succès des essais cliniques » conclut Juliette Pulman, qui se prend même à espérer que cette étape supplémentaire de consolidation sur le modèle organoïde humain pourrait s’étendre à bien d’autres champs pathologies que l’ophtalmologie. On trouve en effet des organoïdes de cerveau, d’intestin, de glandes mammaires… autant de pans de la médecine qui pourraient bénéficier de ces résultats.
Si la technique de vectorisation doit encore être améliorée, afin que plus de cellules répondent à la thérapie génique, la jeune chercheuse envisage avec confiance l’avènement de l’utilisation d’organoïdes dérivés directement de cellules de patients, pour tester des thérapies géniques personnalisées. « Cela ouvrirait la voie à une médecine de précision pour les maladies rétiniennes, où chaque traitement serait adapté au profil génétique du patient », espère Juliette Pulman.
Propos recueillis par Aline Aurias.
Pour aller plus loin : Juliette Pulman, Hugo Malki et al. (2025) Retinal organoids mirror CRISPR Cas9 gene editing efficiency observed in vivo, Molecular Therapy Methods & Clinical Development, Volume 33, Issue 4, 101627.
Pour soutenir la recherche fondamentale sur les maladies de la vision, soutenez l'Institut de la Vision >

À lire aussi

L'anatomie de l'œil
La vue est le plus sollicité de nos 5 sens. Elle nous permet de percevoir les couleurs, les formes, les positions. Elle est essentielle pour intégrer et apprécier le monde qui nous entoure, pour communiquer avec les autres. Admirer ses proches, contempler la nature, observer un tableau, lire un livre, se déplacer, travailler, créer...

Les troubles de la vision et les maladies des yeux de A à Z

Prévention & vision : qui consulter ? A quel âge ?
L’œil est un organe précieux et fragile. Ne dit-on pas de quelque chose à laquelle on est très attaché « j’y tiens comme à la prunelle de mes yeux » ? Il est important de faire contrôler et de préserver ses yeux afin d’acquérir dès le plus jeune âge une bonne vue, et de la conserver toute la vie. L’œil, cette bille de 2,5 cm de diamètre et pesant seulement 7 grammes, abrite pourtant un système très complexe. Directement relié au cerveau, l’œil nous permet de voir et d’interagir avec le monde extérieur.

13 avancées majeures à l'Institut de la Vision
Si le temps de la recherche paraît souvent très long, et tout particulièrement pour les patients atteints de pathologies ne disposant d'aucun traitement, la recherche sur les maladies de la vision a