







Syndrome de Usher, la recherche à grands pas
Avec une prévalence d'environ 1 cas pour 10 000 à 30 000, le syndrome de Usher est une maladie génétique rare, la forme la plus courante de surdicécité combinée. Parce qu'il n'existe pas à ce jour de traitement spécifique, les scientifiques, chercheurs et médecins sont à l'œuvre pour trouver des réponses thérapeutiques adaptées. Cette pathologie a d'ailleurs fait l'objet d'un programme de recherche de grande ampleur, initié par l'Institut de la Vision et l'Institut Pasteur et mobilisant plusieurs équipes de spécialistes sur plusieurs sites. Pour en comprendre la teneur, nous avons fait le point avec le Dr Aziz El-Amraoui, directeur de recherche à l'Institut Pasteur et l'Institut de l'Audition.
Le syndrome de Usher est un déficit neurosensoriel qui associe une perte d’audition ou surdité et des troubles de la vue ou cécité avec, parfois, des troubles de l’équilibre. “C'est un syndrome héréditaire, c'est pourquoi nous avons commencé nos recherches en identifiant d'abord les gènes responsables de la pathologie puis essayé de comprendre pourquoi le défaut dans tel ou tel gène entraînait surdité et cécité chez l'homme. Notre objectif final étant bien sûr de trouver une thérapie pour les patients souffrant de ce syndrome”, précise le Dr El-Amraoui.
Les formes décrites du syndrome de Usher
Trois formes différentes ont déjà été définies : le syndrome de Usher de type 1, une forme qui représente environ 40 % des cas, se traduit par une surdité congénitale profonde non progressive, une déficience sévère de la fonction vestibulaire et une déficience visuelle qui se développe pendant l’enfance. “Rien que pour cette forme, cinq gènes différents ont été identifiés”, précise le directeur de recherche. Moins sévère, le type 2 du syndrome de Usher représente environ 60 % des cas et est caractérisé par une surdité congénitale, moyenne ou sévère, lentement progressive, sans atteinte vestibulaire associée et une rétinite pigmentaire généralement diagnostiquée entre 10 et 40 ans. Enfin, une forme hybride, le syndrome de Usher de type 3, qui représente moins de 3 % des cas et qui s'exprime par une surdité qui survient généralement avant l’âge de 30 ans et progresse à des vitesses variables, associée à une atteinte vestibulaire dans la moitié des cas ; le début de la rétinite pigmentaire survient en général à l’adolescence. Et l'expert d'ajouter : “Nous connaissons aujourd'hui presque l'ensemble des gènes impliqués dans cette pathologie, nous avons également identifié la source de la pathologie Usher, au niveau des cellules sensorielles, les cellules ciliées dans l'oreille et les photorécepteurs dans la rétine.”
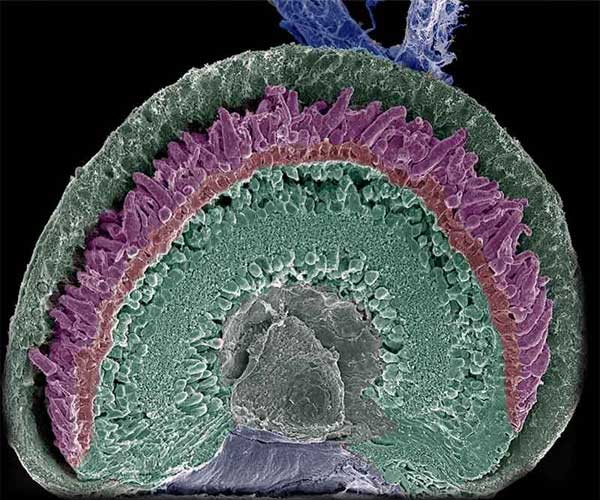
Microscopie électronique à balayage d'un œil d'une larve de xénope, illustrant les différentes couches de la rétine dont la couche des photorécepteurs, colorée enviolet.(Source C. Schietroma, Institut Pasteur).
La piste prometteuse de la thérapie génique
La collaboration entre les équipes de l'Institut de l'Audition et l'Institut de la Vision, notamment celles de Serge Picaud, Deniz Dalkara et José-Alain Sahel, est dans ce projet particulièrement importante. Si, en terme de traitement, lorsque la forme 1 est diagnostiquée, l'implant cochléaire est conseillé afin d'amoindrir l'effet délétère de la perte d'audition, le déficit visuel lui ne dispose à ce jour d'aucune thérapie. “Nous fondons beaucoup d'espoir dans la thérapie génique adaptée au syndrome de Usher, en raison notamment du succès de cette thérapie pour d'autres pathologies, en l'occurrence pour les dystrophies rétiniennes héréditaires associées au gène RPE65”, ajoute le directeur de recherche. De plus, la rétinite pigmentaire chez les patients souffrant du syndrome de Usher apparaît tardivement, au moment de la puberté, laissant ainsi une fenêtre thérapeutique pour intervenir avant que les symptômes ne soient trop sévères.
Des modèles pour reproduire la pathologie Usher au niveau de la rétine
Toutefois, avant d'aller vers des essais cliniques chez l'homme, les chercheurs doivent démontrer sur un modèle animal qu'un traitement fonctionne, et ce en toute sécurité. Or, l'une des difficultés pour le syndrome de Usher vient du modèle souris : la pathologie est parfaitement reproduite au niveau de l'oreille mais elle ne l'est pas au niveau rétinien. “Les molécules du syndrome de Usher sont impliquées dans une structure microvillaire, appelée processus caliciels, qui est très développée dans la rétine de certaines espèces comme les amphibiens, les primates, mais pas chez la souris”, explique le Dr El-Amraoui. Et d'ajouter : “Avec Deniz Dalkara, nous travaillons sur des modèles cellulaires en créant des organoïdes, c'est-à-dire des mini-rétines en 3D, qui ont l'avantage d'être issus de cellules humaines saines et de cellules humaines issues de patients souffrant du syndrome de Usher. L’étude de ces modèles permettra de définir avec précision les étapes critiques de la pathologie à l'échelle cellulaire. Ce travail est complété par l’étude de modèles souris, portant les mêmes mutations, pour obtenir des informations pertinentes à l’échelle de l’organisme entier. Nous essayons parallèlement de créer des modèles chez des espèces qui disposent de cette structure microvillaire, importante pour les études sur le syndrome de Usher. Nous avons commencé chez le xénope (famille des amphibiens) et avons déjà démontré que si nous inactivons un gène Usher, cela entraîne des déformations au niveau des cellules des photorécepteurs, justifiant la perte de vision”, détaille le chercheur. Des projets sont actuellement en cours pour le développement de modèles proches de l'homme chez le grand animal, notamment chez le cochon.
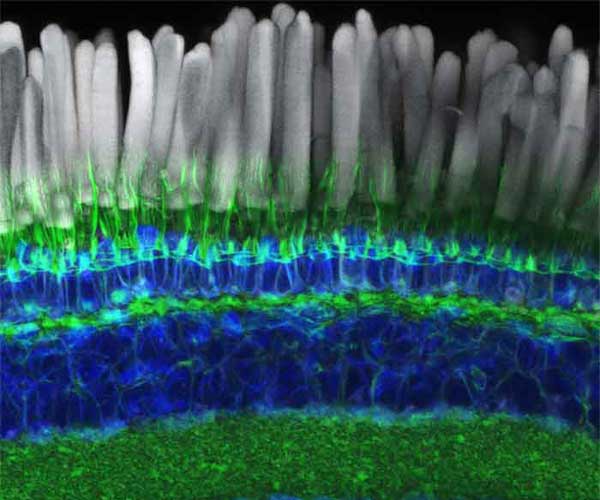
Section de la rétine de xénope montrant un marquage des filaments d’actine (en vert), notamment au sein des processus caliciels qui entourent la base des segments externes des photorécepteurs, le lieu de la phototransduction (marqués en blanc). Les noyaux des cellules sont colorés au DAPI (bleu). (Source C. Schietroma, Institut Pasteur).
Corriger la mutation directement dans le génome
L’approche classique en thérapie génique consiste à insérer une copie fonctionnelle du gène déficient dans le génome. Les travaux en cours chez l’animal montrent que les meilleurs résultats sont obtenus en utilisant pour le transfert de gène dans l’œil, ou dans l’oreille, des virus adéno-associés. Ces vecteurs sont développés pour permettre un transfert contrôlé et une expression efficace et durable du transgène dans les cellules ciblées. “Or la problématique avec le syndrome de Usher, sur les dix gènes identifiés, seuls cinq d'entre eux peuvent - en raison de leur taille - être intégrés dans un virus adéno-associé. Pour les autres gènes, d'autres vecteurs ou d'autres thérapies géniques sont nécessaires pour les corriger”, ajoute le Dr El-Amraoui. Parmi les travaux de recherche qui viennent juste d'être lancés, l'utilisation des “ciseaux à ADN” (CRISPR-Cas9) pourrait contourner ce problème lié à la taille des gènes et est actuellement évaluée dans les modèles pour permettre de corriger la mutation directement au niveau du génome.
En synthèse, les mécanismes du syndrome de Usher ont été clairement identifiés, tant pour l'audition que la vision. La stratégie de thérapie génique adoptée peut être différente et devra être adaptée selon la forme du syndrome de Usher, la taille du gène et/ou la nature de la mutation à l’origine de la pathologie. Pour l'audition, les scientifiques disposent de modèles animaux reproduisant la pathologie Usher et des validations sont en cours pour démontrer les bénéfices de la thérapie génique. Concernant la vision, des efforts sont réalisés pour réussir à reproduire sur des modèles animaux adaptés la pathologie humaine au niveau de la rétine. En parallèle sont menées des recherches sur des organoïdes rétiniens, permettant de reproduire la pathologie dans un contexte génomique et cellulaire humain et ainsi évaluer la thérapie génique comme une réponse aux atteintes de ce syndrome.
Le projet de recherche LIGHT4DEAF
Initié par les Prs Christine Petit, directrice de l'Institut de l'Audition, et José-Alain Sahel, directeur de l’IHU Foresight, le projet de recherche hospitalo-universitaire LIGHT4DEAF a vu le jour en 2015. Il a mobilisé durant cinq ans une équipe multidisciplinaire de scientifiques, médecins et professionnels de santé dans un effort commun visant à comprendre la maladie, développer des traitements innovants et informer les malades et les acteurs impliqués dans la prise en charge de la maladie. L’Institut de la Vision, l’Institut Pasteur, le CHNO des Quinze-Vingts, le service d’ophtalmologie et de médecine physique et de réadaptation de l’hôpital de la Pitié Salpêtrière, les services ORL de l’hôpital Necker Enfants malades, de l’hôpital de la Pitié Salpêtrière et de l’hôpital Robert Debré, l’Université Clermont-Auvergne se sont investis dans ce projet. L’ensemble des découvertes ont été présentées en octobre 2021 au Usher Info Scientific Symposium.

La collaboration entre les équipes de l'Institut de l'Audition et l'Institut de la Vision est particulièrement importante dans ce projet visant à mieux comprendre le syndrome de Usher et développer des traitements spécifiques. En photo : Serge Picaud et Deniz Dalkara de l'Institut de la Vision. © CNRS

Dr Aziz El-Amraoui, directeur de recherche à l'Institut Pasteur et l'Institut de l'Audition
Pour soutenir l'Hôpital Necker-Enfants malades, rendez-vous sur leur site>

À lire aussi

Pour soutenir la recherche sur les maladies de la vision, focus sur l’Institut de la Vision-Paris, centre de recherche international

WebTV Institut de la Vision : "la médecine personnalisée"
Alors que nombre d’affections oculaires sont désormais connues et reconnues, que les progrès diagnostiques et thérapeutiques en ophtalmologie sont considérables, la très grande variabilité inter-ind
Douleur oculaire, une prise en charge unique au CHNO des Quinze-Vingts
La sécheresse oculaire est l’un des premiers motifs de consultation dans les cabinets d’ophtalmologie&nbs