







Rétinopathie pigmentaire, l'actualité recherche

Un nouveau gène impliqué dans une forme rare de rétinopathie pigmentaire liée au chromosome X vient d’être identifié. Une découverte qui ouvre de nouvelles perspectives pour le diagnostic et le développement de traitements ciblés. Entretien avec les chercheuses à l’origine de cette découverte.

Isabelle Audo, Médecin chercheuse spécialisée dans les maladies rétiniennes génétiques, professeure en ophtalmologie à Sorbonne Université, directrice adjointe de l’Institut de la Vision et coordinatrice du Centre de référence Maladies Rares à l’hôpital des 15-20.

Christina Zeitz, Directrice de recherche Inserm. Chimiste, généticienne. Codirectrice de l’équipe "Identification de défauts génétiques conduisant à des maladies oculaires progressives ou non progressives" à l’Institut de la Vision avec la Pr Audo et responsable du Département Génétique.
La rétinopathie pigmentaire, encore appelée dystrophie de type bâtonnet-cône, est une maladie génétique dégénérative qui touche les photorécepteurs, les cellules de la vision situées dans la rétine. Pathologie rare, concernant environ une personne sur quatre mille, elle touche indifféremment les personnes des deux sexes, quel que soit leur groupe ethnique, et peut débuter à tout âge, même si l’incidence d’apparition est plus importante entre 10 et 30 ans.
Une pathologie qui mène progressivement à la cécité
Cette pathologie dégénérative touche d’abord les bâtonnets, ce qui se manifeste par une perte progressive de la vision nocturne ou en faible intensité lumineuse, et des difficultés d’adaptation à l’obscurité, lors du passage d’une pièce éclairée vers une pièce sombre. Les cônes se retrouvent ensuite affectés, ce qui mène à un rétrécissement du champ visuel périphérique, qui provoque une vision « en tunnel ».
"A l’exception de certaines formes peu sévères ou d’apparition tardive, la rétinopathie pigmentaire progresse la plupart du temps vers la cécité."
L'atteinte est bilatérale et, les deux yeux se trouvant affectés, va avoir un impact important sur la qualité de vie quotidienne : incapacité à se diriger à la nuit tombée, ou dans les endroits mal éclairés, maladresse, collision avec des objets ou des personnes situés en périphérie du champ visuel, interdiction légale de la conduite automobile... Plus tardivement, des troubles de la vision de certaines couleurs (un phénomène nommé dyschromatopsie), en particulier le bleu et le jaune, ainsi qu’une sensibilité accrue aux fortes intensités lumineuses (photophobie) peuvent également apparaître.
Si la vision centrale est conservée jusqu’à des stades tardifs de la maladie, elle finit également par être affectée dans les formes les plus sévères, avec une diminution progressive de l’acuité visuelle, empêchant de lire ou d’effectuer des tâches minutieuses. A l’exception de certaines formes peu sévères ou d’apparition tardive, la pathologie progresse la plupart du temps vers la cécité. Il n’existe à l’heure actuelle qu’un seul traitement approuvé, le Luxturna, qui ne concerne que 0,3 à 1% des cas associés à des mutations sur le gène RPE65. Plusieurs autres essais cliniques de thérapie génique sont cependant en cours pour cibler d’autres gènes, voire agir indépendamment des mutations génétiques, dont l’objectif est de ralentir voire stopper la progression de la dégénérescence rétinienne en cause.

Pr Isabelle Audo - Institut de la Vision © Institut de la Vision – Cyril-Bruneau
De nombreux variants impliqués
Suspectée en cas de mauvaise vision nocturne ou d’antécédents familiaux, la rétinopathie pigmentaire est diagnostiquée par l’examen du fond d’œil, qui montre, dans les formes avérées, des amas pigmentés noirs, et confirmée grâce à une méthode appelée électrorétinogramme, qui analyse les réponses électriques des photorécepteurs à une stimulation lumineuse. D’origine génétique, la rétinopathie pigmentaire est donc liée à des mutations de gènes codant pour des protéines importantes pour le bon fonctionnement des photorécepteurs ou de leur couche de soutien, l’épithélium pigmenté rétinien.
A l’heure actuelle, plus d’une cinquantaine de gènes ont été identifiés, avec des prévalences très différentes, comme associés à la rétinopathie pigmentaire. Le profil d’expression des gènes associés est variable avec certains gènes exprimés également dans d’autres tissus que la rétine, dont les mutations ne donnent qu’une atteinte isolée à la rétine ou d’autres responsables d’un syndrome tel par exemple celui de Usher.
Selon les gènes mutés, la rétinopathie pigmentaire va se transmettre selon le mode autosomique dominant (un seul gène muté suffit à provoquer la maladie), autosomique récessif (il faut avoir reçu un allèle muté de chacun de ses deux parents), ou liée au chromosome X. Ces dernières, qui touchent principalement les hommes, représentent 15% des cas de rétinopathie pigmentaire et sont généralement celles qui présentent le phénotype le plus sévère et les débuts les plus précoces.
"Seuls trois défauts génétiques avaient jusqu’à présent été identifiés comme responsables de rétinopathies pigmentaires liées à l’X."
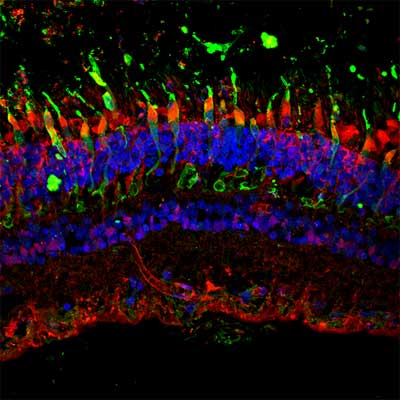
Marquage par immunofluorescence de la localisation de la protéine IDH3G (en rouge) dans les photorécepteurs (ici en vert) sur une coupe de rétine humaine. © Institut de la Vision
Un nouveau gène identifié sur l’X
Jusqu’à présent, seuls trois défauts génétiques avaient été identifiés comme responsables de rétinopathies pigmentaires liées à l’X. Mais de nombreux cas inexpliqués, et dont l’histoire familiale semblait pointer vers une transmission liée à l’X, laissaient penser que d’autres gènes étaient probablement impliqués. C’est l’un de ces gènes que la Pr Isabelle Audo et la directrice de recherche Christina Zeitz, codirectrices de l’équipe « Identification de défauts génétiques conduisant à des maladies oculaires progressives ou non progressives » à l'Institut de la Vision, ont récemment identifié. « En général, on suspecte une maladie liée à l'X quand les mères ne sont pas atteintes, mais peuvent présenter des signes très distinctifs au fond d'œil, alors que leurs pères, leurs oncles peuvent être atteints, et que leurs fils le sont », expliquent les chercheuses.
Epaulées par Lorenzo Bianco, étudiant en médecine italien venu passer un an à leur côté, les deux scientifiques ont analysé le génome et identifié, chez quatre patients non apparentés, quatre mutations sur le même gène susceptibles de causer une rétinopathie pigmentaire liée à l’X. En effet, IDH3G code pour une des trois sous- unités d’une enzyme, appelée isocitrate déshydrogénase mitochondriale (IDH3), qui est impliquée dans l’un des mécanismes de production d’énergie dans nos cellules, le cycle de l'acide citrique, aussi appelé cycle de Krebs. Cette enzyme est exprimée dans les segments internes des photorécepteurs, ce qui en fait un bon gène candidat pour une affection touchant ces cellules rétiniennes. De plus, des variants de IDH3A et IDH3B, codant pour les autres sous-unités de IDH3, ont déjà été associés à une rétinopathie pigmentaire autosomique récessive non syndromique.

"La pathologie liée à la mutation de IDH3G progresse moins rapidement que les autres rétinopathies pigmentaires liées à l’X, ce qui pourrait permettre d’envisager plus facilement des traitements."
Un mécanisme encore à élucider…
De façon étonnante, bien que les trois protéines constituant l’IDH3 soient exprimées dans d’autres tissus que la rétine, leur mutation ne semble pas provoquer d’autres atteintes phénotypiques que la rétinopathie pigmentaire. Plusieurs hypothèses peuvent être avancées pour expliquer cela : « C'est probablement lié au fait que les variants identifiés ne mènent pas à une absence totale de protéines. Les photorécepteurs étant parmi les cellules du corps les plus gourmandes en énergie, une telle diminution pourrait impacter leur fonctionnement sans pour autant perturber celui du reste de l’organisme. Mais on peut également envisager que, hors de la rétine, les autres organes expriment des protéines à la fonction redondante de celle d’IDH3, et qui prendraient donc le relais, gommant son défaut de production dans les autres tissus » précise Isabelle Audo.
… mais des impacts déjà concrets
Si la compréhension des conséquences exactes de ces mutations, et des possibles solutions thérapeutiques, est encore à venir, l’identification des variants va déjà permettre d’intégrer ce gène dans la liste des gènes à analyser lors du diagnostic d’une rétinopathie pigmentaire. Il sera ainsi possible d’apporter à certains patients une première forme de réponse sur la cause de leur pathologie. Un point non négligeable également sur le plan du conseil génétique en vue d’un projet parental. « Nous pensons que la découverte de nouveaux gènes est très importante pour les patients, pour qu’ils gardent l’espoir qu’un jour on comprenne ce qui cause leur pathologie, même si cela ne débouche pas tout de suite sur un traitement » soulignent les deux collègues.
De plus, le phénotype lié à IDH3G étant moins sévère que la plupart des autres types de rétinopathie pigmentaire liée à l’X, cette variante de la pathologie pourrait permettre d’envisager plus facilement des traitements que certaines formes dans laquelle la dégénérescence est beaucoup plus rapide. « Par rapport aux personnes présentant une mutation sur les autres gènes représentant la majorité des formes liées à l’X, où les patients sont quasiment aveugles quand ils atteignent 30 à 40 ans, chez les patients que nous avons identifiés il y a une préservation de la structure rétinienne centrale avec une vision utile qui fait qu’au même âge ils sont encore capables de lire, d'écrire… » décrit Christina Zeitz. La fenêtre thérapeutique pour prendre en charge les patients mutés sur IDH3G est donc plutôt large et pourrait permettre de tenter des approches correctives (thérapie génique par complémentation ou remplacement de gènes, technologie CRISPR, approches pharmacologiques) avant la dégénérescence complète des bâtonnets puis des cônes.
Pour soutenir la recherche fondamentale sur les maladies de la vision, soutenez l'Institut de la Vision >

À lire aussi

Les troubles de la vision et les maladies des yeux de A à Z

13 avancées majeures à l'Institut de la Vision
Si le temps de la recherche paraît souvent très long, et tout particulièrement pour les patients atteints de pathologies ne disposant d'aucun traitement, la recherche sur les maladies de la vision a
Lipides et rétinopathie diabétique : une avancée vers un traitement précoce
Recherche sur le diabète et ses risques pour la santé des yeux : deuxième partie de notre interview auprès de Xavier Guillonneau, chercheur Inserm à l'Institut de la Vision.

Mieux comprendre la malvoyance
Il y a quelques années la notion de malvoyance était mal connue. Les personnes déclarées en cécité légale, définie par un seuil de performance visuelle ouvrant droit à une prise en charge orthoptique, étaient considérées comme non voyantes.